There has been a great deal of conversation in the genetic genealogy community over the past couple of weeks about the use of “small” segments of matching DNA. Typically, the term “small” refers to segments of 5 cM and smaller, although some people include segments of 7 cM or even 10 cM and smaller in the definition.
The question, essentially, is whether small segments of DNA can be used as genealogical evidence, and if so, how they can be used.
While it may seem at first that all shared segments of DNA could constitute genealogical evidence, unfortunately some small segments are IBS, creating “false positive” matches for reasons other than recent ancestry. These segments sometimes match because of lack of phasing, phasing errors, or a variety of other reasons. One thing, however, is clear: there is no debate in the genetic genealogy community that many small segments are false positive matches. There IS debate, however, regarding the rate of false positive matches, and what that means for the use of small segments as genealogical evidence.
Small Segments and Me
Because small segments are prone to be false positives, I personally take a very cautious approach. For example, when I download my list of shared segments from the Family Tree DNA, the very first thing I do with the spreadsheet is sort it by size and delete every segment smaller than 5 cM. For me, there is too much uncertainty surrounding small segments to base any conclusion on them.
While this may not be the answer for everyone, it is the responsibility of the genealogist to place a VERY high burden on any argument that utilizes these small segments, if only because of the known high rate of false positives. As we’ll see, a “maybe” is not good enough for a genealogist using DNA evidence in a proof argument.
What is the Rate of False Positives?
Because large databases of genomic data are still relatively new, most IBD studies have been performed using simulated data. In a study earlier this year using real genomes (including thousands of genetic genealogists) scientists from 23andMe examined IBD segments in a group of 25,000 individuals which included 3,000 father-mother-child triads.
In the study, a segment of DNA shared by a child with someone in the database other than their parents was declared to be IBD if that same segment of DNA was shared with a parent (importantly, the IBD segments only had to overlap by 80%, not a full 100%).
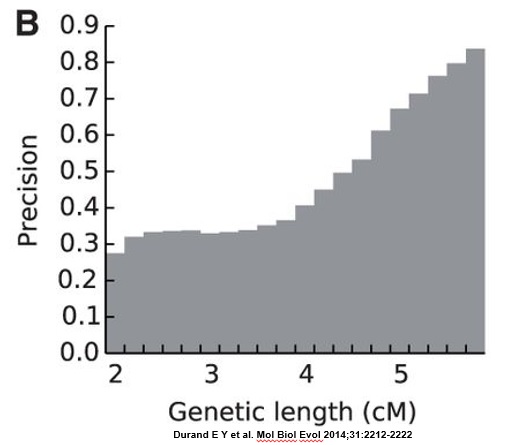
The researchers found that more than 67% of all reported segments shorter than 4 cM are false-positive segments (see FIG. 2B, below). At least 60% of 4cM segments were false-positive, and at least 33% of 5 cM segments were false-positive. The number of false-positives decreased fairly rapidly above 5 cM.
To my knowledge, this study is the largest of its kind to examine this question directly. The paper is available online for free (http://mbe.oxfordjournals.org/content/31/8/2212).
EDIT (4 December 2014) – Ann Turner has noted that the genomes used to generate the graph above were phased using the popular BEAGLE phasing program. The segment data we get from 23andMe and Family Tree DNA, on the other hand, is based on unphased genomes. This means that the false positive error rate for that segment data is almost certainly higher – and potentially MUCH higher – than what is reported in the chart above.
As support for this conclusion, John Walden has shared IBD v. IBS numbers from a private study (see http://www.isogg.org/wiki/Identical_by_descent). Now I provide these numbers only because so much of the community is familiar with them. However, because these numbers have never been peer-reviewed and the methodology has never been satisfactorily shared, I strongly recommend use of the 23andMe study above instead. That being said, here is what Walden found in his study, in which he examined whether segments found in children were (IBD) or were not (IBS) found in the child’s parents:
| cM | % IBD | % IBS |
| 10 | 99 | 1 |
| 9 | 80 | 20 |
| 8 | 50 | 50 |
| 7 | 30 | 70 |
| 6 | 20 | 80 |
| 5 | 5 | 95 |
According to Walden’s study, 95% of 5 cM segments were false-positive segments (not found in the parents). Again, I would take Walden’s numbers with a grain of salt. I feel much more confident in the large-scale 23andMe study, however.
So until another study is published, there can be little debate that the false positive rate for segments of 5 cM or smaller is likely somewhere between 33% and 95%.
If segments of 5 cM and smaller are largely or mostly false positives, how can we use these in our research? Is there any way to salvage these smaller segments?
Parent-Child Data
Perhaps the only way, currently, to use small segments is to compare the small segments with immediate family members (parents), and even this method is extremely limited. The theory here is that if I share a small segment with a cousin, and my mother shares the same segment with the same cousin, then I inherited that segment from my mother and it is most likely a real segment (rather than IBS). But note that this theory is not ironclad!
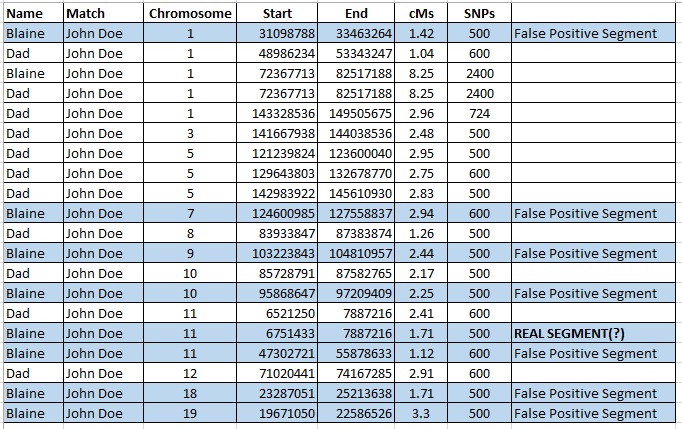
The table below is segment data from Family Tree DNA for me and my father, showing where we each share segments of DNA with a genetic cousin, John Doe. All of my small segments are highlighted in blue. Of course, my father and I each share one “large” segment of DNA with John Doe, an 8.25 cM segment on Chromosome 1. All other segments that I share with John Doe are 3.3 cM or smaller.
When I compare these small segments to my father’s results, I see that of my eight small segments, only one (12.5%) is shared by John Doe, me, AND my father (my mother doesn’t match John Doe although theoretically I could have inherited one or more of these segments from her). The segment that John Doe, my father, and I share in common is very small, 1.71 cMs, and I do wonder if that could be the result of a “pile-up” or some other false positive state.
This process does not, however, safely extend to more distant family members and cousins, for example if my parents were not able to test. After all, John Doe is a cousin, and yet I share many, many false positive segments with him. I’m not certain how far I would comfortably extend this method.
Out of curiosity, I wondered how many other people I shared some of these small segments with, and whether my father shared that segment with any of those matches.
So, I found the first 1.42 cM false positive segment in a sorted spreadsheet of all my matches:
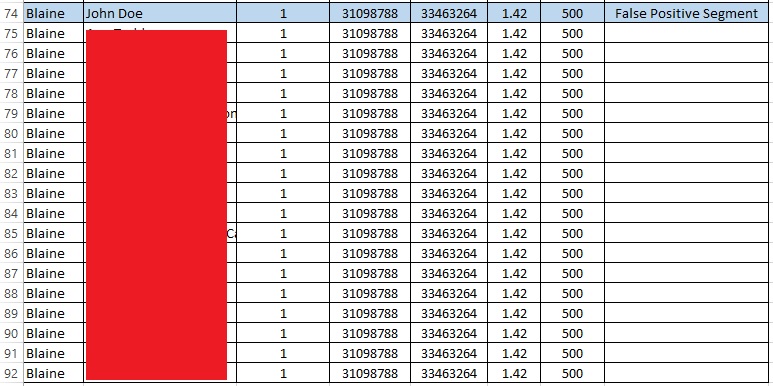
The list goes on and on like this; in fact, I share that same exact small segment with 48 other people. My father shares the segment with 2 people (not me), neither of whom match me. (My mother shares the segment with 19 people, 8 of whom share the segment with me). Therefore, my parents share this segment with 21 people, and I share it with 48 people (and only 8 of those people share the segment with me and with a parent, my mother). There is clearly something very wrong with this segment, but I only learned that by conducting this intensive analysis (that I normally would not do!).
In contrast, look at the “real” small segment. The sharing is much more limited, and John Doe is the only person that both my father and I match at that segment.
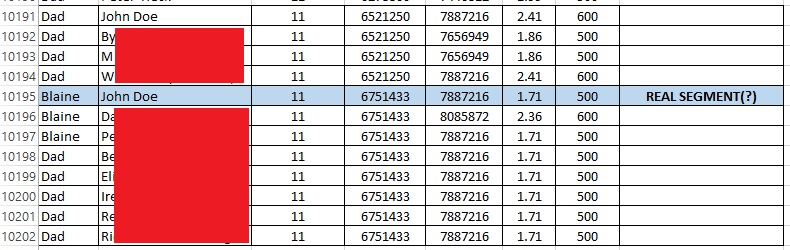
I do feel a bit better about this particular segment, but even so I could not use this segment alone as evidence (instead I’d rely on the larger segment). For the reasons in the next section I would be concerned about using this “real” small segment rather than relying on larger segments.
So why isn’t the parent method ironclad? Because sharing of a small segment between a parent and a child does not guarantee that the small segment is IBD. The first and foremost reason that these “confirmed” segments could still be a false positive for matching is that the segment could be shared by many members of a population or region (i.e., ancestry outside the genealogically-relevant timeframe), rather than just by closely related cousins. If many members of a population share a segment in common, it’s very easy to confuse that for close relatedness if you’re only looking at a few members. So, as an extreme example, if 75% of people from Upstate New York all carry a 5 cM segment that conveys increased shivering ability (hey, it’s cold up here), then if I test two Upstate New Yorkers it’s going to look like they have recent ancestry with a 5 cM segment, when in fact their common ancestor lived 500 years ago.
The Larger Segment Theory
I have had people tell me that if they share a large segment of DNA with a group of triangulated cousins, and they also share a small segment with the same group of triangulated cousins, then the small segment must be real. Taken to an extreme, it has been argued that people that match those “confirmed” small segments (whether or not they match the large segment) can triangulate to the same family group. Charted out, it might look something like this:
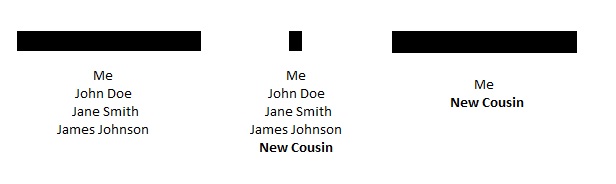
However, there are several real concerns with this, even if there were two or more of the small segments:
- New Cousin could be related through an entirely different line of the family, especially if this New Cousin comes from a similar time and place;
- The small segment sharing with New Cousin could be a false positive. Although more small segments (and/or more matching cousins) may decrease this likelihood, even 2 or 3 small shared segments like this could conceivably be pure chance;
- The segment could be from a “pile-up”, meaning that the small segment is widespread throughout a particular population and is not indicative of recent ancestry;
- And there may be more; I’d be interested to have others chime in here.
Thus, this use of small segments is especially problematic.
The hypothesis above could only work with larger segments, for example if New Cousin and James Johnson shared a large segment. But if that were the case, your argument would be based entirely on large segments and could avoid the problems associated with small segments.
Using Small Segments as Evidence?
So can small segments ever be utilized as evidence in a genealogical proof?
Well, whenever a proof argument uses a small segment as evidence – EVEN IF THE SMALL SEGMENT IS IN CONJUNCTION WITH LARGER SEGMENTS – scrutinize the information VERY closely and ask at least the following questions:
- Is the small segment shared by parents of BOTH matches? If there is no parent to test, or the answer is no, then you should probably discard the small segment. And even if it is shared by the parent, question whether it could be shared for another reason. Further, keep in mind that even if it is a “real” segment that I share with my parent, that is not a guarantee that it is a “real” segment in my match; maybe it is an IBS for them, not shared with their parent!
- How many of your other matches share the small segment? This is less clear, but the more people that share the small segment in your match list, the more concerned I would be, particularly if there is no known relationship.
There is no doubt that many small segments are indeed real, but the genetic genealogist’s ability to decipher between a “real” small segment and a “false” small segment is extremely limited and suspect. Ultimately I think it’s possible to identify segments that are probably real, but currently it is nearly impossible to use them in a meaningful way to support a genealogical hypothesis.
I’m very interested to hear your thoughts. How can we find ways to utilize these small segments? Are they hopeless, or is there a light at the end of the tunnel?
.
Thank you, the best thing I’ve ever read on the subject 😉
As for the light at the end of the tunnel re the use of small segments in autosomal genetic genealogy, I’m thinking the hope is in rare SNP’s, touchy subject that they are.
And, if you step outside the genetic genealogy community and read scientific genetic literature, you’ll find I’m not the only one saying that.
Thank you Laura, I’m glad you enjoyed it! I think rare SNPs could indeed be part of the answer, as will phasing and the identification/elimination of small segments involved in “pile-ups.” I think there’s lots of light at the end of the tunnel!
Here is the problem with your conclusions. If small matches were that likely to be false positive, then statistically one would expect to find them randomly in population groups. That is, I should have as many false positives in Germany as in Scotland, or as many from New York as in Georgia. But I don’t. I find my proportions in each place to be precisely what my family tree would suggest – overwhelmingly from Scotland and Georgia, very few from New York and Germany. That is, I find virtually all of my so-called ‘false positives’ precisely where I would expect my distant relations to be located geographically. Statistically, that would seem extraordinarily unlikely. I think the science needs to be reconsidered.
Stan – your conclusions sound solid at first glance, but you’re forgetting a VERY important point about the data. Your match list (and thus all shared segments of DNA) is intentionally BIASED toward actual genealogical relatives that came from the places you would expect. Every single genetic match in your match list shares a long segment of DNA with you, and most of these matches are genealogical relatives. Thus, if your genealogical relatives came from Scotland and Georgia, then many of your genetic matches will be from Scotland and Georgia, and thus the small segments will look like they came from Scotland and Georgia.
In contrast, if you upload your raw data to GEDmatch and lower the matching threshold to 1cM, you will match nearly everyone the database, from all over the world. In other words, only using this method will you “find them randomly in population groups” around the world (heavily BIASED by who in the world has tested, of course).
Blaine
Blaine – a great article!! I think you are right on. Our problem is that there is no good way for us to determine if even a triangulated small segment is IBD or IBS/IBC. To me it seems that a matching algorithm, with ACTGs from both parents available for each SNP, can put together what appear to be matching segments in the short run. This is an analysis that needs to be done – how many shared segments (of different sizes) actually match phased data? In other words, how large a segment can an algorithm construct that doesn’t really match true phased data. This thinking comes to me from chromosome mapping, where most segment ends overlap a little. Ckearly the algorithm doesn’t know exactly where to start or end, because segments from different ancestors don’t overlap. Depending on the outcome of such an analysis, I think stacking, or pile up, comes from a unique combination of ACTGs on both sides that lend themselves to matching small segments (perhaps in addition to the endogamy theory that there are common, small, DNA pieces in our DNA soup from distant ancestors – another oft used statement that seems reasonable, but has little actual data for it). Thanks again, Blaine, for honing in on this issue.
Thank you Jim, that means a lot coming from you! Imagine if everyone received their phased data from AncestryDNA with the pile-up regions identified? That would be incredibly valuable.
I wouldn’t delete the segments under 5 cM. What I do is map the matching segments even if I think there is a good chance it is IBS as long as there are other, larger segment matches from the same participant. So, if I matched a participant named Bettinger with segments of 20 cM, 4.9 cM, and a bunch of stuff 2 cM and below, I would label the long one on the chromosome map as “Bettinger” and the next one I would label “Bettinger (b)”. If I have other matches to the same (“b”) spot, then I see if they follow some identifiable genealogical pattern. If I get enough of them, I conclude that it is there for a reason. If not, then eventually it will get erased from the chromosome map due to other, more reliable segment matches. But I put all the matches on the map (more or less), knowing that I will be doing a lot of erasing. The map evolves with new data, or data that I am just now getting to analyze.
As a practical matter, I have plenty of longer matches to deal with anyway, so will only get to those shorter, “b” segments when they appear as part of a larger whole that demands attention. These things happen rarely, but I feel rarity is still better than zero, which is what you get by deleting those under 5 cM by rule.
Gregg
P.S. I am the “child” in a TPOCT with phased data all around. Sometimes it is phased data to phased data comparison, where I am very confident at less than 5 cM.
Gregg – I think that is a good method, and it sounds like you’re making it work. I couldn’t agree more about the longer matches; they keep me so busy I don’t know if I could ever get to the smaller matches!
Although you have a great grasp of the limitations of small segments, I don’t think the community as a whole – especially newbies – understand them.
Thank you for reading!
Gregg, even though I’m fairly new in using DNA segments for genealogical research, I agree with your use of small segments. Just this week I used the GEDmatch Tier 1 Matching Segment Search for the first time and got a 60+K line .csv file. After downloading this as a spreadsheet and sorting by chromosome, start position, and cM, I started visually scanning down through the spread sheet and noticed that in a number of places on all chromosomes i would see clumps of small matches that had identical starting positions, and often indentical stopping positions. As I looked further, I saw that in many cases, above these small segments, I’d find much larger samples from others with identical, or sometimes earlier, starting positions. I would then often find those same people lumped together on the same or other chromosomes. Since that observation I been trying to see if I can work up some way to automate gathering more data to see if this will help me figure out which ancestor(s) I might connect with which position on which chromosome. Have you seen similar results? Blaine, if you see and read this, what do you think?
Thank you so much for this article! While I have a basic understanding of genetic genealogy, I fall over when trying to apply it in a practical way. This was a very understandable explanation and I will save it for reference until it is pounded into my head!
Kathleen – I’m so glad you enjoyed it! Thank you for reading!
Glad to see you sound this cautionary note. The danger of making unsupported leaps is just as real in genetic genealogy as it is in traditional research. It’s all too easy to “ass-u-me” when we really, really want to prove a connection.
The requirement for evidence and proper citations is so important! Thank you for stopping by!
Thanks for the article – I am still trying to absorb the whole triangulation/chromosome browser concept. I am still not clear about how close the “overlapping” of the segments should be? for example, above you state that the segment 6751433 7887216 is a match. How far off can one of the numbers be before it would not be a match? for example, if the numbers were 6741255 7887216 would that be considered a match? Can you clarify you sentence “…(importantly, the IBD segments only had to overlap by 80%, not he full 100%).” Is this related to my question? How do you determine if a segment overlaps by 80 or 100%? Which overlaps which and does it matter? Sorry if I am being obtuse? Kind of new to this.
Thanks
David – there is no perfect answer to your question, and it may be answered differently depending on who you ask. The Durand paper I reference above only required an overlap of 80%, so the numbers you propose above would indeed be considered a match (at least it looks like they would by eyeballing them, I didn’t actually do the math, I’ll let you do that!). Determining the overlap is as simple as lining up the two segments and determining if there is at least 80% overlap between them.
Keep in mind, this 80% overlap is an arbitrary number chosen by Durand. They could have chosen 90%, or 75%, or something else.
Dr. Ann Turner pointed out on the that Durand et al. study used phased data and that without phasing our small segments have a higher rate of false positives. In the report they say they used Beagle to make detection of all pairwise IBD segments in a cohort of over 100,000 individuals computationally feasible with GERMLINE.
Armando – I saw Ann’s comment, and she is absolutely correct. Thank you for sharing it with me nonetheless!
What that means, in reality, is that for 23andMe and Family Tree DNA, the false positive rate will be even HIGHER than 67% (AncestryDNA phases data before matching)! I don’t believe it will reach the 95% that Walden predicts, but then again, who knows; I’m basing that on some VERY limited and anecdotal evidence.
Thank you again, excellent point!
Thank you for a great article and for alerting us to the important research by Durand et al. published in MBE.
The Durand et al study research addresses the probability of a single DNA segment being IBS rather than IBD. However, I believe most of the genetic genealogist who advocate exploring small segments caution against drawing conclusions based on one small segment. Instead, they recommend looking for matches on several segments AND only when there is some other reason for expecting a relationship. The Durand et al. study provides some useful information on error rates which allows us to calculate the probability that a match on several small segments. As noted in the study, the probability of one matching DNA segment being a false positive (IBS) is .67. Thus, the probability of two small matching segments being false positives is .45 (.67 x .67). The probabilities decline as the number of matching segments increases: 3 (.30), 4 (.20), 5 (.14), 6 (.09), 7 (.06), 8 (.04), 9 (.03), 10 (.02), etc. Thus, even with ten matching segments, there is a possibility that the match is a false positive but the probability is only 2% – in other words, there is a 98% probability that the match is a true positive (IBD). Note that even single large segments (above 7 cM) carry a small probability that the match is not IBD. The question, then, is what level of probability one feels comfortable with in concluding there is probably a relationship. For example, with four small matching segments there is a 80% probability the match is IBD but with six matching segments the probability increases to 91%.
Durand et al. indicate that they have developed a program which can detect false positives more directly, rather than through probabilistic models. They call the program HaploScore and it can be downloaded from https://github.com/23andMe/ibd. That might be an alternative light at the end of the tunnel. .
Willi – (see my comment to you below, and I’ll add this). First, the Durand paper significantly underestimates the false positive rate, since it was using PHASED data. We’re all using unphased segment data from 23andMe and FTDNA. Second, as CeCe Moore’s recent blog post showed, Durand’s method of IBD detection – declaring a segment as IBD if it is shared by both a parent and a child with another person – is faulty. See: http://www.yourgeneticgenealogist.com/2014/12/the-folly-of-using-small-segments-as.html
That doesn’t mean there isn’t light at the end of the tunnel, just that it’s a teeny bit dimmer! Thanks again for reading and taking the time to leave a great comment, much appreciated!
I find it instructive to look at the distribution of segment numbers in Family Finder matches. If the small segments are contributing support to identifying the MRCA, I’d expect the number of segments to decrease with more distant relationships, since we inherit less and less DNA with each successive generation.
For me, the average number of segments in the different relationship bins doesn’t change much at all:
3rd to 5th: 11.9 segments
4th to distant: 11.5 segments
5th to distant: 11.4 segments
To me, that’s an indication that it’s too easy to construct short segments just by coincidence. They’re what I call pseudo-segments in my JoGG column “Identity Crisis.”
http://www.jogg.info/72/files/Turner.htm
Note: this article was written several years ago, and I might propose a different method to study these segments now that we can do a batch download of Chromosome Browser segments.
I made another observation in the course of generating numbers for this post. I have one outlier, a person who shares just two segments with me. He carries an African mtDNA haplogroup, while my background is European. I suspect this is another example of how it’s easy to construct short segments if you belong to the same general gene pool with similar allele frequencies. But it’s harder for African Americans, and FTDNA’s methodology may impact their ability to identify cousins.
Ann – I’m slowly working on another way to analyze the numbers myself, and I really, really like your point here. Did you use a program to determine these numbers? If you don’t mind, I will include an excerpt from this comment in my new post on the subject. I’ll contact you for permission. Thank you for taking the time!
Willi – that’s right, and many of us have been hoping that 23andMe would be utilizing HaploScore to reduce false positive segments (and matching). So far, they haven’t used it publicly. There are several lights at the end of the tunnel, thank goodness!
Blaine, after taking my time and reading this several times (and skipping the Facebook discussion), I think I understand all the points you are making.
But your Upstate New York example still accepts that there was some common ancestor sometime in the past and a person doesn’t get even a small segment just by breathing the same air as the general population.
At GRIP, I accepted the logic that small segments are not worth the trouble, without accepting that they are totally and absolutely irrelevant.
I wrote in my blog a couple of days ago, that I consider it perfectly reasonable to accept that a small segment shared by me, my sisters, my aunt and my uncle would also be shared by my late father if we could test him. And further it would also be reasonable to attribute that segment to one of my grandparents (or shared between them.)
But I also said “Whether that segment came down unchanged and intact from one of my great-grandparents can be a matter of legitimate debate. Whether it came unchanged and intact from someone two-three generations further back can be a matter of legitimate doubt.”
I would not use it for “new” relatives, but then I am not in this for the new relatives.
That policy still seems right to me and not in conflict with anything you write above.
Israel – regarding my Upstate New York example, I do indeed believe that some small fraction of the “real” small segments – including possibly those infamous “pile-ups” – are due to ancient shared ancestry. Whether those are useful for genealogical purposes is another question. I think most of them are not, as they really only indicate a shared ethnicity rather than a recent common ancestor (while “a shared ethnicity” is also due to common ancestry, it’s due to ancient shared ancestry).
I don’t think your policy is unreasonable. A small segment shared by at least 5 extremely close family members at two different generations indicates to me that your father and likely your grandfather/grandmother possessed that small segment. Sharing by siblings AND children would be too coincidental. Beyond your grandparents’ generation, as you point out, no one knows. And as you wisely point out, I wouldn’t use it at all for finding or confirming “new” relatives.
I think you’re using just the right balance of inquiry/logic and skepticism. Kudos.
Regarding “pileups”. You are addressing smaller segments, but if the segments are more than 10cms, can the word “pileup” be used. E.g., there are more than 50 of us at Gedmatch who overlap on a with more than 10 cMs, and the “leader of the group” says these large segments are a “pileup”. I believe they are real matches. Please comment.
Sorry to be slightly off-point.
Caith – the honest answer is that “no one knows.” We don’t know how long the pile-up segments at AncestryDNA can get. 10 cM doesn’t seem exceedingly big to me to be a pile-up. And the fact that 50 people at GEDmatch, which has a relatively small database, match at the same suspect makes me very suspicious of the validity.
A thought re: the idea that a father and son having the same small segment makes that segment more useful for identifying a match. The fact that one has inherited the segment from one’s father does not affect the liklihood that the segment IN THE FATHER is IBD to the matching segment in the hypothesized relative (unless we accept sticky segments). Since we have the father’s results in this case, we can use his DNA to consider relative matches. He would be more closely related to cousins from his side, hence more useful. The fact that the son did or did not inherit some segment would add no information. This line of thought suggests that knowing a father and son share a small segment would not tell us anything about the usefulness of the segment.
On the basis of this article, I have a slightly bigger question: If on such very limited example and such a small in scale experiment, DNA analysis can give so much false positives, how can one be so sure about the validity of DNA analysis for the larger picture (both in the Y-DNA and mt-DNA lineages for humans) and the DNA based evidence for an “out-of-Africa” theory?
I see you don’t use the potential of social websites like facebook on your blog.
You can get huge traffic from social sites on autopilot using one useful tool,
for more info search in google for:
Alufi’s Social Automation
Deleting segments under 5cM seems like an extreme. I have two known fourth cousins who are brother and sister. John appears as a cousin on FTDNA sharing 12.7 cM. Sue does not appear on either FTDNA or Ancestry as a cousin. When I compare with their Gedmatch kits, set at 1 cM and 500 SNPs, I share 74 cM with John and 40 cM with Sue. The largest segment shared with Sue is 4.5 cM. When I compare all three kits, we have 17.3 cM in common over 8 segments. Had I deleted segments under 5 cM, and not known that Sue was a cousin, I would not have found her using only segments over 5cM.
The 12.7 cM triangulated segment with John came from our 5th gr-grandfather, so it’s probable that any IBD segments under 5 cM came from a 7th-9th gr-grandparent. It’s true that not many of our trees are complete back 11 or 12 generations, so we may have difficulty assigning matching segments to a CA. Our present inability to assign them might change with one cousin’s build out of his tree. Hopefully you are deleting these small segments into deep storage where they might be retrieved as our abilities, both technical and genealogical improve.
I’m presently checking the one half of my matches with Sue that didn’t match her brother. The temptation is to call them IBS, but as they include our two largest matches (over 4 cM), it will be interesting if they match other relatives on that family line.
Hopefully the under 5cMs do not mean much, because I share 4.9 cMs with my husband. LOL
Sometimes people forget to check the X chromo to add to the totality.
I have also had interest in small matching segments. I am member of Gedmatch and routinely set CM default to 1 CM for all one-to-one comparisons with people I show one-to-many matches with default parameter 7 CM.
Found a very interesting pattern at one Chromosome. Approximately half of matches that I know are of Irish ancestry I have been finding a match of between 1.1 to 2.3 CMs on Chromosome 6 from approximately 25,000,000 to 34,000,000. This morning I tallied number matches on Chromosome 6 in this segment out of total one-to-one comparisons I have recently done. The total comparisons were 36. The total number of matches on Chromosome 6 were 18. One other very strange characteristic of these matches were the consistently very high SNP at there segments. For smaller matches on this segment (1.1-1.2 CM) the SNP is between 1-2 thousand. For larger matches up to 2.3 the SNPs are up to several thousand.
I had read of false positives. One site suggested checking archaic matches and compare on Gedmatch one-to-one at 1 CM default parameter. Gist of article was if find match on same chromosomal segment likely to be false positive. I did this check and none of archaic matches were at Chromsome 6 from 25…… to 34……
I have not compared this segment to my children’s autosomal results. All four of my children had DNA tested. Will do that next. Do not have a parent who has had DNA checked. Will have to wait for grandchildren, test them and then see of three generation incidence is found, as per Ce Ce Moore article you referenced.
For what it is worth, I have one grandparent who was French-Canadian. Other three were of Irish descent. One grandparents ancestors migrated to New Brunswick Canada in first half of 10th Century from Armagh, Cork and other places. These ancestors settled for 2-3 generations in western New Brunswick and Aroostook County Maine (extreme northeastern Maine) along upper St. John River.
The other lines were from southern Galway with migration in early 20th Century to Massachusetts and New York, and Sligo-Mayo with migration in latter 19th century to Pennsylvania coal country. My YDNA is M222. Father from southern Galway (Kilthomas parish).
If this interests you then you have my permission to share with others. I will answer e-mails.
That is strange, or a coincidence. I have recently noticed a lot of triangulated matches where we all share small overlapping segments on this area of chromosome 6. I am new to genealogy so I don’t know what it means. I thought it might mean there is a common ancestor in a certain area of the UK. One of the matches involved is a close relative, another somebody who we both share a bigger segment of DNA with, and the others who I share a bigger segment of DNA with and my close relative shares just this area on chromosome 6 with them.
Footnote.
Checked same group one-to-one comparisons with two of my children. Results showed high percentage of recurrence of chromosome 6 segment in both children. Recurrence rate was less than for myself. Recurrences of chromosome 6 segment were not identical for each child. One child was 10 out of 36. Other child was 9 out of 36. Both had chromosome 6 segment on 8 of 36.
I am not sure I agree with your conclusions First of all they are all on our individual DNA from small to large segments I personally lower the threshold down to 1 and change nothing else I can then see the full range of matching segments . I also use other factors such as surname s , family connections , time zones , geographical areas archeological aspects rather than just maths and physics hypothesis to massage the results I would have missed some highly important connections if I followed your conclusions Your article is well expressed but it is something I don’t accept Sorry You gave personal examples to support it but I could also give personal examples to support me . There are a number of experts in the subject but I would just advise people to use common sense and use other factors too Alan
“For example, when I download my list of shared segments from the Family Tree DNA, the very first thing I do with the spreadsheet is sort it by size and delete every segment smaller than 5 cM”
You’re missing a lot then.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083094/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3350316/
I’m ignorant about DNA. However, I would appreciate your opinion on the use of matching small 4-6 cM segments (~200-300 SNP) among known 6-8th cousins with a potential 8th cousin about whom we are trying to determine if he lies along the same family line? For instance, on GEDMatch I find 6 (known to be distant cousins) out of 7 match at one particular chromosome segment at this level with the possible 8th cousin at this level. Seems unlikely these would all be false positives. I don’t know about phasing (all parents are deceased). Is it worthwhile to recruit more known distant cousins for this study? If they to match on this segment, that would be encouraging. I really want to arrive at a probability of a valid common ancestor to us all. I’ve been told that RefinedIBD software won’t help (for larger populations).
to continue … suppose I do one-to-ones with another 20 randomly selected GEDMatch kit numbers and find say only 10% match on this segment. If the known cousins match at a 90% clip, then I would guess (I’m sure too naively) that the probability of a kinship with the 8th cousin would then be 90%. It can’t be this simple, right?
Get your matching segments now. It’s pretty easy. Hop onto for various DNA kits, reviews and amazing deals they are offering here. https://www.dnatestreview.org/
A great post once again, as is usually expected from this website. For those interested in SNPs, I’ve come to find that https://www.dnatestreview.org/ offers tests based on SNPs and is also a great platform to read reviews on various kits and avail excellent deals.